Gas Separation
We computationally explore phenomena related to gas separation by employing a combination of quantum chemical methods and classical simulation techniques. For this purpose we develop force-fields from first principles to be used in classical simulations.
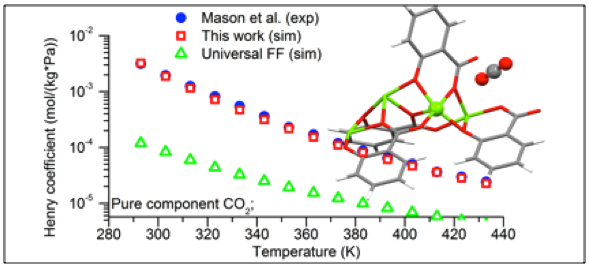
Performance of different force field in predicting gas adsorption in MOFs.
Reference:
A. L. Dzubak, L.-C. Lin, J. Kim, J. A. Swisher, R. Poloni, S. N. Maximoff, B. Smit & L. Gagliardi, Ab initio carbon capture in open-site metal-organic frameworks,
Nature Chemistry, 4, 2012, pp 810-816
DOI: 10.1038/nchem.1432