Quantum Chemical Methods Development
We explore multireference methods based on the active space formalism and find ways to simplify them and make them affordable for realistic systems. We also combine multireference methods with density functional theory with the aim of being able to study middle-to-large size systems at an affordable cost. We are currently working on Multiconfiguration Pair-Density Functional Theory (MC-PDFT), a generalization of DFT based on multiconfigurational wave functions. MC-PDFT is especially useful for multireference systems including those containing transition metals, excited states, transition states, and other open-shell systems not well described by a single Slater determinant.
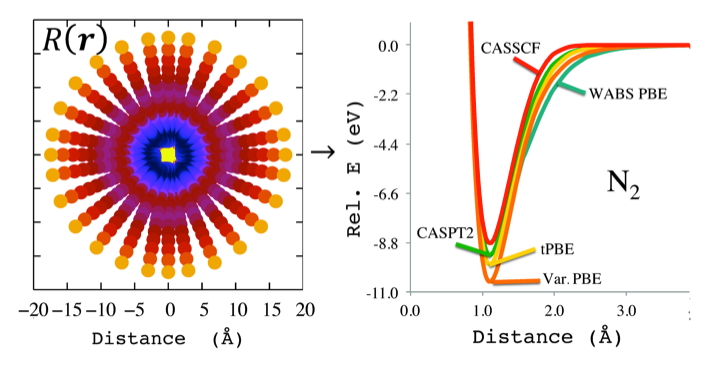
Plot of a quantity related to the on-top pair density for the N2 molecule
Reference:
G. Li Manni, R. K. Carlson, S. Luo, D. Ma, J. Olsen, D. G. Truhlar, and L. Gagliardi, Multi-Configuration Pair-Density Functional Theory, J. Chem. Theory Comput., 10 (9), 2014, pp 3669-3690
DOI: 10.1021/ct500483t
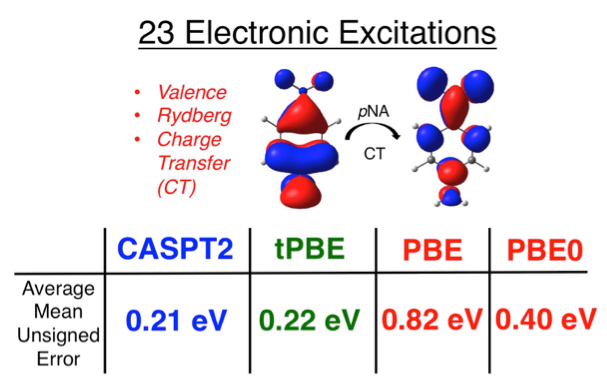
Examples of electronic excitations investigated with various electronic structure methods
Reference:
C. E. Hoyer, S. Ghosh, D. G. Truhlar, and L. Gagliardi, Multiconfiguration Pair-Density Function Is as Accurate as CASPT2 for Electronic Excitation, J. Phys. Chem. Lett., 7, 2016, pp 586-591.
DOI: 10.1021/jacs.5b12515